
Sickle Cell Anemia: September is Sickle Cell Awareness Month
by Beverly Schaefer, MD
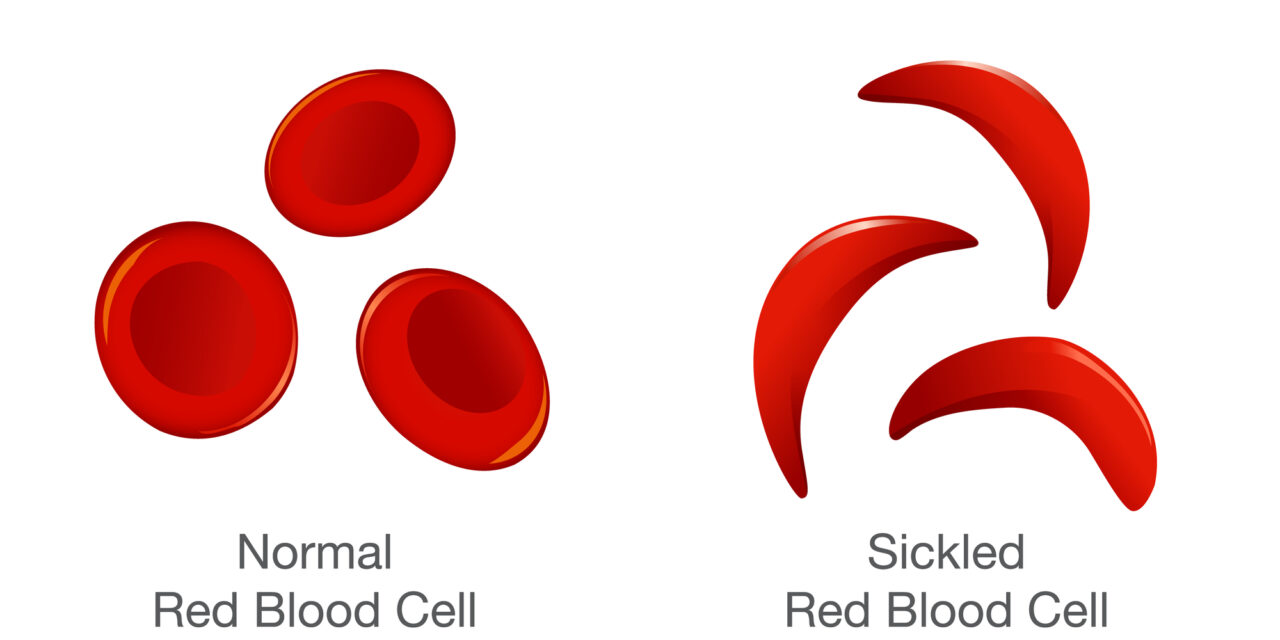
Sickle cell disease (SCD) affects roughly 100,000 people in the U.S. Sickle cell anemia is a genetic disorder that affects the red blood cells. Normal red blood cells are round and smooth, and move through blood vessels easily. Sickle cells are crescent-shaped and become hard and sticky, often getting stuck in small blood vessels, blocking blood flow and causing pain and organ damage. Because sickle cells die quickly, the body cannot make new red blood cells to replace them fast enough, resulting in anemia.
SCD is an inherited, lifelong disease, acquired by inheriting two genes for sickle hemoglobin, one from each parent. Individuals most at risk come from Africa, South or Central America, the Caribbean Islands, Mediterranean countries, India, and Saudi Arabia. People with only one sickle cell gene are said to have the sickle cell “trait.” They do not have the disease, but can pass the gene to their children. In the U.S., about 1 in 13 African Americans has the sickle cell trait.
SCD is one of the most powerful examples of health care disparities in this country. SCD patients experience worse health outcomes compared to other diseases, and have access to fewer health resources. Most patients are African American Medicaid beneficiaries, and less than 70% of doctors accept new Medicaid patients. Health providers also often inaccurately perceive SCD patients as drug-seekers, often doubting the severity of their pain, resulting in much longer wait times to receive care and relief. A shortage of physicians experienced in caring for these individuals compounds the problem.
The average life expectancy for people with severe SCD is 30 years shorter than for those without SCD. The rate of stroke in African American adults ages 35-64 with SCD is three times higher than rates in African Americans of similar age without SCD, and patients with SCD have the highest rate of returning to the hospital within 30 days of being discharged. They typically experience significant pain, infections, acute chest syndrome, and other complications, including vision loss or blindness, leg ulcers, stroke, deep vein thrombosis, pulmonary embolism, organ damage, kidney disease, impotence, and pregnancy complications.
There are treatments for SCD that can significantly decrease complications. Traditionally, a cure was only possible with a bone marrow transplant, which takes healthy cells of blood from one person (typically a sibling) and puts them into someone whose bone marrow is not working properly. However, in recent years, gene therapy by means of gene addition or gene editing (using CRISPR) may hold significant promise and possible cure for individuals with sickle cell disease who do not have a bone marrow donor. These therapies are currently being studied, and are not yet FDA approved, but there is hope on the horizon for patients living with this disease.
Beverly Schaefer, MD is an Assistant Professor of Pediatrics, Pediatric Hematology and Oncology, practicing at UBMD Pediatrics, Oishei Children’s Hospital, Roswell Park, and WNY Blood Care. See https://medicine.buffalo.edu/faculty/profile.html?ubit=bschaefe.








{kind=link}